Introduction
Breast cancer remains a leading cause of cancer-related mortality worldwide, with molecular subtype classification playing a critical role in treatment selection and patient outcomes. Traditional histopathological approaches, while valuable, often lack the molecular resolution needed to capture the full spectrum of tumor heterogeneity. Modern bioinformatics and machine learning offer transformative potential by enabling comprehensive analysis of genomic data to both understand cancer biology and automate clinical decision-making.
This project presents an integrated multi-modal framework combining RNA sequencing analysis, co-expression network biology, and machine learning to classify breast cancer subtypes—HER2-positive, Triple-Negative (TNBC), Non-TNBC, and normal tissue. The work was executed as a collaboration between complementary expertise: bioinformatician Samuel Adegbola led the RNA-seq pipeline (quality control, alignment, differential expression, and WGCNA network analysis), while machine learning development led by myself focused on dimensionality reduction and predictive modeling for automated classification.
The complete pipeline progresses from raw FASTQ files through differential expression analysis (DESeq2), co-expression network construction (WGCNA), functional enrichment (GO/KEGG/Reactome), and culminates in a Support Vector Machine classifier achieving 88.33% accuracy. This end-to-end approach not only identifies individual biomarkers and coordinated gene modules but translates these discoveries into a deployable clinical tool, demonstrating the power of integrating systems biology with machine learning for precision oncology.
Phase 1: PROJECT SETUP AND INITIAL DATA HANDLING
Step 1: Create a Cloud Storage Bucket
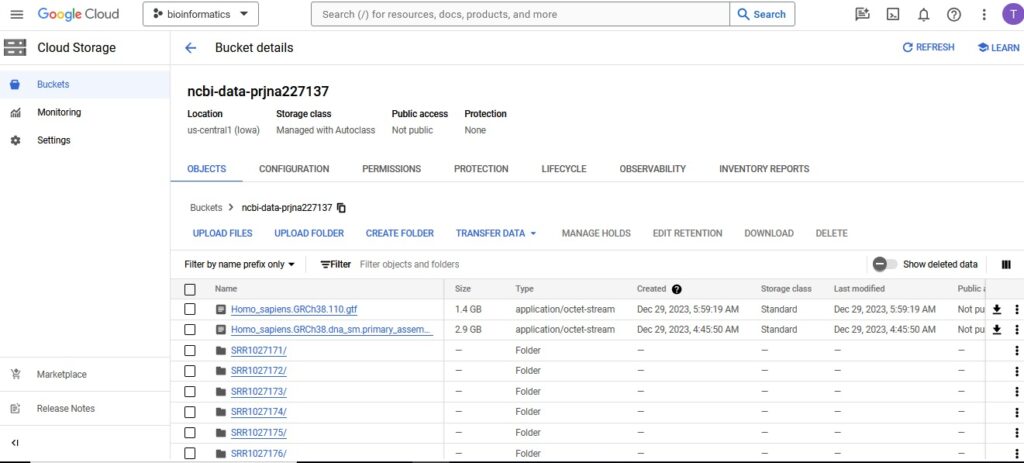
The first step was to create a Cloud Storage bucket named ncbi-data-prjna227137 to store all project data.
gsutil mb gs://ncbi-data-prjna227137
Step 2: Data Collection
Next, data was collected from the public NCBI dataset (GSE52194) and stored it in the Cloud Storage bucket. Using NCBI’s SRA Run Selector service, patient metadata and FASTQ files was then downloaded directly to Cloud Storage by granting temporary Storage Object Admin access to NCBI’s service account. 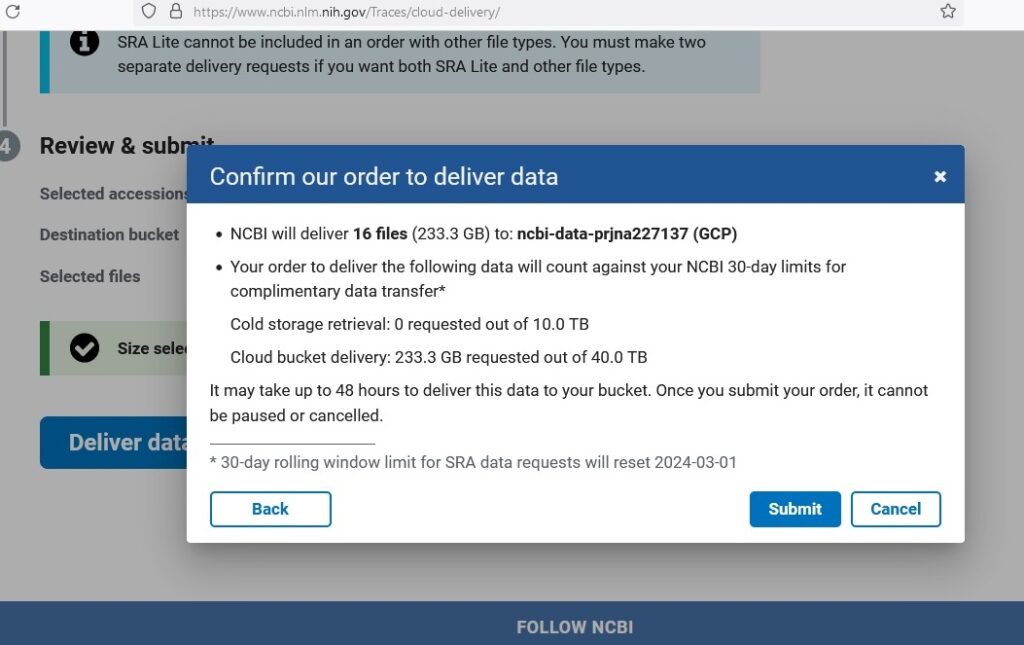
Phase 2: DATA PREPROCESSING AND FEATURE SELECTION
Step 3: Build and Upload a Container Image
Docker containers were built for RNA sequence analysis. Below is the Dockerfile that was used:
FROM continuumio/miniconda3
RUN apt-get update && apt-get install -y \
procps \
cutadapt \
fastqc \
rsem \
bwa \
samtools \
bcftools
The Docker image was built and run:
docker build -t my_rna_seq_image -f Dockerfile .
docker run -it --name my_rna_seq_container my_rna_seq_image /bin/bash
Due to dependency issues with trim-galore, I created a new conda environment with Python 3.9:
conda create -n new_env python=3.9
conda activate new_env
conda install -c bioconda trim-galore
Step 4: Downloading the Reference Genome and Annotation Files
For this project, the necessary reference genome and annotation files from Ensembl was obtained using the following wget command. This ensured that there is most up-to-date and accurate data for human genome analysis.
wget -P /rna-seq-folder/ https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa.gz
wget -P /rna-seq-folder/ https://ftp.ensembl.org/pub/release-112/gtf/homo_sapiens/Homo_sapiens.GRCh38.110.gtf.gz
Unzip the downloaded files:
gunzip /rna-seq-folder/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa.gz
gunzip /rna-seq-folder/Homo_sapiens.GRCh38.110.gtf.gz
Step 5: Preparing the Reference for RSEM
With the reference genome and annotation files downloaded and unzipped, the following command was executed to prepare the reference:
rsem-prepare-reference --gtf /rna-seq-folder/Homo_sapiens.GRCh38.110.gtf --star /rna-seq-folder/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa /rna-seq-folder/ref/human_rsem_ref
Step 6: Automate Data Processing
A script was written to automate downloading, quality checking, and trimming RNA-Seq data:
#!/bin/bash
start=1027171
end=1027190
for ((i=start; i<=end; i++))
do
bucket_path="gs://ncbi-data-prjna227137/SRR$i/"
gsutil -m cp "${bucket_path}*" .
fastqc SRR${i}/*sequence.txt.gz
trim_galore --quality 20 --fastqc --paired SRR${i}/*_1_sequence.txt.gz SRR${i}/*_2_sequence.txt.gz
gsutil cp SRR${i}/*trimmed.fq.gz "${bucket_path}"
done
The script was made executable and run:
chmod +x process_rna_seq.sh
./process_rna_seq.sh
This phase involved setting up a robust framework for the project and ensuring the initial data handling processes were thorough and well-documented. The quality control, read alignment, and expression quantification steps laid a solid foundation for subsequent phases of the project.
Phase 3: DATA INTEGRATION AND ANALYSIS
Step 7: Align and Estimate Expression with RSEM
RSEM was run to align reads and estimate gene and isoform expressions using the following script:
#!/bin/bash
cd /rna-seq-folder/fastq
for file in *_1_sequence.txt.gz; do
base=$(basename $file _1_sequence.txt.gz)
read1=${base}_1_sequence.txt.gz
read2=${base}_2_sequence.txt.gz
rsem-calculate-expression --paired-end --star --num-threads 16 \
$read1 $read2 \
/rna-seq-folder/ref/human_rsem_ref \
/rna-seq-folder/results/${base}
done
The script was made executable and run:
chmod +x run_rsem.sh
./run_rsem.sh
Step 8: Convert RNA-Seq Output to CSV
The .results files were converted to .csv format using the following script:
#!/bin/bash
for file in *.results; do
newfile="${file%.results}.csv"
awk 'BEGIN{FS="\t";OFS=","} {gsub(/ /, "\",\"", $2); print "\"" $1 "\",\"" $2 "\",\"" $3 "\",\"" $4 "\",\"" $5 "\",\"" $6 "\",\"" $7 "\""}' "$file" > "$newfile"
echo "Converted $file to $newfile"
doneThe script was made executable and run:
chmod +x convert_results_to_csv.sh
./convert_results_to_csv.shStep 9: Combining Data in Google Colab
After preparing the RNA-Seq output data, multiple CSV files needed to be combined laterally to create a matrix format suitable for DESeq2 analysis. Here’s how it was done:
Step 1: Set Up the Environment – The pandas library was ensured to be available for data manipulation.
Step 2: Upload Files to Google Colab – In Colab, the CSV files were uploaded to the session storage using the sidebar.
Step 3: Import Pandas – Pandas was imported in the notebook:
!pip install pandas
import pandas as pd
import osStep 10: Read and Combine Data Horizontally
A script was written to read each CSV file and merge them horizontally based on the gene_id column:
# Initialize an empty DataFrame or the first DataFrame
key_df = None
# List all files and merge them horizontally
for filename in os.listdir('/content'):
if filename.endswith('.csv'):
# Read the CSV file
data = pd.read_csv('/content/' + filename)
# Extract the sample identifier from the filename
sample_id = filename.replace('.csv', '')
# Select and rename the necessary columns
data = data[['gene_id', 'expected_count']].rename(columns={'expected_count': sample_id})
if key_df is None:
key_df = data
else:
# Merge while avoiding duplicate column names
key_df = key_df.merge(data, on='gene_id', how='outer')
# Handle any missing values or discrepancies
key_df.fillna(0, inplace=True)
print(key_df.head())
# Save the merged DataFrame to a CSV file
output_file_path = '/content/merged_data.csv'
key_df.to_csv(output_file_path, index=False)
print(f"Merged data has been saved to {output_file_path}")Phase 4: DIFFERENTIAL EXPRESSION ANALYSIS
Step 11: Set Up DESeq2 in R Environment and Run DESeq2 Analysis
DESeq2 and other necessary packages were installed, the libraries were loaded, and the DESeq2 environment was set up. The count data was loaded and metadata for the dataset was created. The metadata includes the condition for each sample (Normal, HER2, NonTNBC, TNBC). A DESeq2 dataset was then created using the count data and metadata, and the DESeq2 analysis was run to identify differentially expressed genes.
# Install and load necessary packages
install.packages("pheatmap")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("biomaRt")
BiocManager::install("DESeq2")
BiocManager::install("clusterProfiler")
BiocManager::install("org.Hs.eg.db")
BiocManager::install("ReactomePA")
library(pheatmap)
library(biomaRt)
library(DESeq2)
library(clusterProfiler)
library(org.Hs.eg.db)
library(ReactomePA)
# Load count data (replace the file path with your actual path)
count_data <- read.csv("/content/count_data.csv", row.names = 1)
# Create metadata for the full dataset
metadata <- data.frame(
row.names = colnames(count_data),
condition = c(rep("Normal", 3), rep("HER2", 5), rep("NonTNBC", 6), rep("TNBC", 5))
)
# Convert non-integer values to integers by rounding
count_data <- round(count_data)
# Create DESeq2 dataset for the full dataset
dds <- DESeqDataSetFromMatrix(countData = count_data, colData = metadata, design = ~ condition)
# Pre-filtering (Optional) Remove rows with low counts to improve computation
dds <- dds[rowSums(counts(dds)) > 1, ]
# Normalize the data and run DESeq2 analysis
dds <- DESeq(dds)
# Extract normalized counts
normalized_counts <- counts(dds, normalized = TRUE)Step 12: Differential Expression Analysis
Differential expression analysis was performed for each condition compared to Normal. Significance thresholds were defined and significant results were extracted (adjusted p-value < 0.05, log2FoldChange > 1). The top differentially expressed genes for each comparison were identified.
# Perform differential expression analysis for each condition vs Normal
res_her2_vs_normal <- results(dds, contrast = c("condition", "HER2", "Normal"))
res_non_tnbc_vs_normal <- results(dds, contrast = c("condition", "NonTNBC", "Normal"))
res_tnbc_vs_normal <- results(dds, contrast = c("condition", "TNBC", "Normal"))
# Define significance thresholds for heatmap, Volcano, & MA plot
padj_threshold <- 0.05
log2fc_threshold <- 1
# Extract significant results (adjusted p-value < 0.05, log2FoldChange > 1)
sig_HER2_vs_Normal <- subset(res_her2_vs_normal, padj < padj_threshold & abs(log2FoldChange) > log2fc_threshold)
sig_NonTNBC_vs_Normal <- subset(res_non_tnbc_vs_normal, padj < padj_threshold & abs(log2FoldChange) > log2fc_threshold)
sig_TNBC_vs_Normal <- subset(res_tnbc_vs_normal, padj < padj_threshold & abs(log2FoldChange) > log2fc_threshold)
# Top genes for each comparison
top_genes_HER2 <- rownames(head(sig_HER2_vs_Normal[order(sig_HER2_vs_Normal$padj), ], 50))
top_genes_NonTNBC <- rownames(head(sig_NonTNBC_vs_Normal[order(sig_NonTNBC_vs_Normal$padj), ], 50))
top_genes_TNBC <- rownames(head(sig_TNBC_vs_Normal[order(sig_TNBC_vs_Normal$padj), ], 50))Step 13: Convert Ensembl IDs to Gene Symbols and Entrez IDs
Ensembl IDs were converted to gene symbols and Entrez IDs using the biomaRt package.
# Convert Ensembl IDs to Gene Symbols and Entrez IDs
mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
convert_ids <- function(ensembl_ids) {
ids <- getBM(filters = "ensembl_gene_id",
attributes = c("ensembl_gene_id", "hgnc_symbol", "entrezgene_id"),
values = ensembl_ids,
mart = mart)
ids <- ids[match(ensembl_ids, ids$ensembl_gene_id), ] # Ensure the order matches the input
ids$hgnc_symbol[is.na(ids$hgnc_symbol)] <- ids$ensembl_gene_id[is.na(ids$hgnc_symbol)]
return(ids)
}
# Convert top genes to gene symbols and Entrez IDs
top_genes_HER2_ids <- convert_ids(top_genes_HER2)
top_genes_NonTNBC_ids <- convert_ids(top_genes_NonTNBC)
top_genes_TNBC_ids <- convert_ids(top_genes_TNBC)Step 14: Save Significant Results
Gene symbols were added to significant results and saved to CSV files.
# Add gene symbols to significant results
sig_HER2_vs_Normal$gene_symbol <- convert_ids(rownames(sig_HER2_vs_Normal))$hgnc_symbol
sig_NonTNBC_vs_Normal$gene_symbol <- convert_ids(rownames(sig_NonTNBC_vs_Normal))$hgnc_symbol
sig_TNBC_vs_Normal$gene_symbol <- convert_ids(rownames(sig_TNBC_vs_Normal))$hgnc_symbol
# Reorder columns to have gene_symbol first
sig_HER2_vs_Normal <- sig_HER2_vs_Normal[, c("gene_symbol", names(sig_HER2_vs_Normal)[-ncol(sig_HER2_vs_Normal)])]
sig_NonTNBC_vs_Normal <- sig_NonTNBC_vs_Normal[, c("gene_symbol", names(sig_NonTNBC_vs_Normal)[-ncol(sig_NonTNBC_vs_Normal)])]
sig_TNBC_vs_Normal <- sig_TNBC_vs_Normal[, c("gene_symbol", names(sig_TNBC_vs_Normal)[-ncol(sig_TNBC_vs_Normal)])]
# Sort significant results by adjusted p-value
sig_HER2_vs_Normal <- sig_HER2_vs_Normal[order(sig_HER2_vs_Normal$padj), ]
sig_NonTNBC_vs_Normal <- sig_NonTNBC_vs_Normal[order(sig_NonTNBC_vs_Normal$padj), ]
sig_TNBC_vs_Normal <- sig_TNBC_vs_Normal[order(sig_TNBC_vs_Normal$padj), ]
# Save significant results to CSV files with gene symbols as rownames
write.csv(sig_HER2_vs_Normal, "sig_her2_vs_normal.csv", row.names = FALSE)
write.csv(sig_NonTNBC_vs_Normal, "sig_non_tnbc_vs_normal.csv", row.names = FALSE)
write.csv(sig_TNBC_vs_Normal, "sig_tnbc_vs_normal.csv", row.names = FALSE)Top 20 Differentially Expressed Genes (HER2 vs Normal)
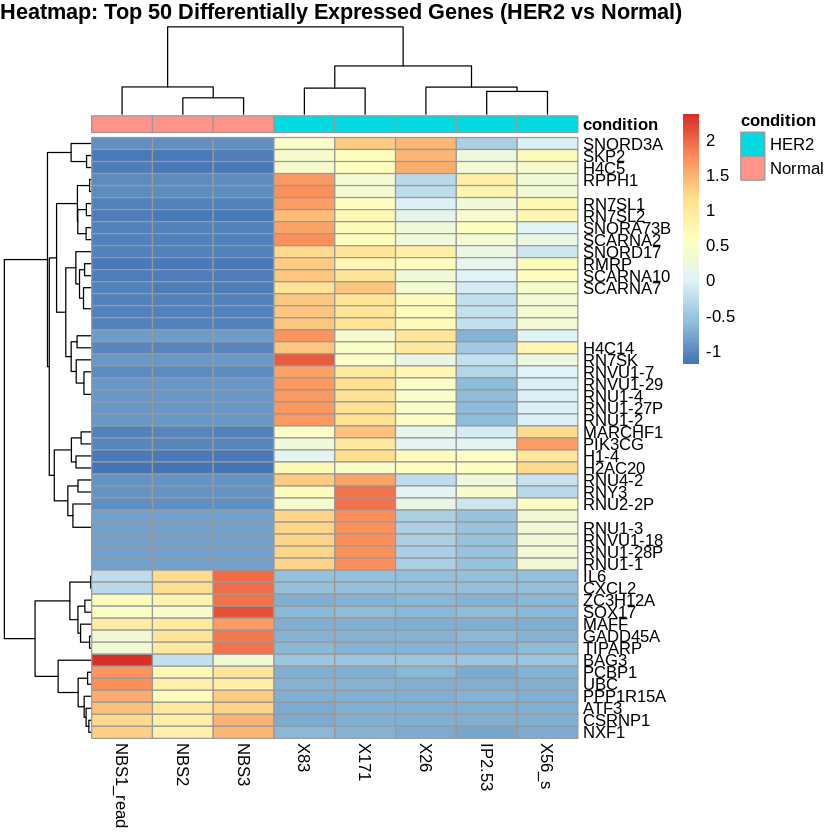
| gene_symbol.hgnc | log2FoldChange | pvalue | padj |
|---|---|---|---|
| RPPH1 | 13.120665 | 4.82e-125 | 1.70e-120 |
| RN7SL1 | 10.489661 | 5.37e-119 | 9.46e-115 |
| RMRP | 11.149963 | 2.34e-109 | 2.74e-105 |
| SCARNA7 | 9.912687 | 1.50e-87 | 1.32e-83 |
| RN7SL2 | 9.188408 | 5.38e-85 | 3.79e-81 |
| SNORD3A | 11.934661 | 4.36e-82 | 2.56e-78 |
| RNU1-27P | 14.122484 | 5.41e-80 | 1.90e-76 |
| RNU1-2 | 14.122484 | 5.41e-80 | 1.90e-76 |
| RNU1-4 | 14.122484 | 5.41e-80 | 1.90e-76 |
| RNVU1-29 | 14.122484 | 5.41e-80 | 1.90e-76 |
| 10.763495 | 1.20e-76 | 3.83e-73 | |
| SNORA73B | 12.135523 | 3.70e-76 | 1.08e-72 |
| SCARNA2 | 9.716538 | 9.39e-75 | 2.54e-71 |
| RN7SK | 10.376260 | 2.32e-71 | 5.84e-68 |
| PPP1R15A | -7.007382 | 9.80e-66 | 2.30e-62 |
| MAFF | -7.113582 | 8.15e-64 | 1.79e-60 |
| 13.666196 | 9.85e-53 | 2.04e-49 | |
| H1-4 | 8.177815 | 4.96e-51 | 9.71e-48 |
| SKP2 | 4.212157 | 1.93e-50 | 3.57e-47 |
| GADD45A | -6.132037 | 3.91e-49 | 6.88e-46 |
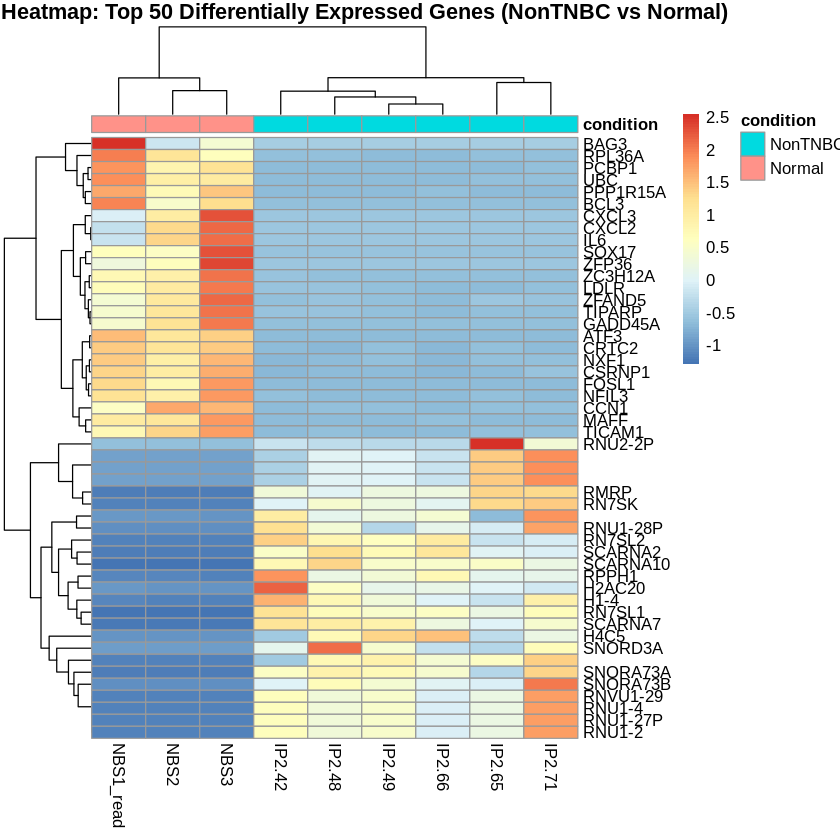
Top 20 Differentially Expressed Genes (NonTNBC vs Normal)
| gene_symbol.hgnc | log2FoldChange | pvalue | padj |
|---|---|---|---|
| RPPH1 | 13.337359 | 1.57e-136 | 5.37e-132 |
| RN7SL1 | 10.061270 | 8.76e-117 | 1.50e-112 |
| RMRP | 10.242729 | 2.14e-98 | 2.45e-94 |
| RN7SL2 | 9.581003 | 2.94e-98 | 2.52e-94 |
| SCARNA2 | 10.275627 | 1.04e-87 | 7.10e-84 |
| SCARNA7 | 9.645241 | 4.28e-86 | 2.44e-82 |
| SNORA73B | 11.588326 | 4.75e-72 | 2.33e-68 |
| SNORD3A | 10.433383 | 4.33e-67 | 1.86e-63 |
| MAFF | -6.885642 | 4.42e-65 | 1.68e-61 |
| PCBP1 | -3.491532 | 9.62e-65 | 3.30e-61 |
| RN7SK | 9.428783 | 4.64e-63 | 1.44e-59 |
| PPP1R15A | -6.360018 | 1.22e-58 | 3.29e-55 |
| 9.102245 | 1.25e-58 | 3.29e-55 | |
| RPL36A | -4.799584 | 5.54e-56 | 1.36e-52 |
| RNU1-27P | 11.463050 | 1.97e-55 | 3.75e-52 |
| RNU1-2 | 11.463050 | 1.97e-55 | 3.75e-52 |
| RNU1-4 | 11.463050 | 1.97e-55 | 3.75e-52 |
| RNVU1-29 | 11.463050 | 1.97e-55 | 3.75e-52 |
| UBC | -5.645960 | 4.05e-55 | 7.31e-52 |
| TIPARP | -4.146750 | 1.83e-52 | 3.13e-49 |
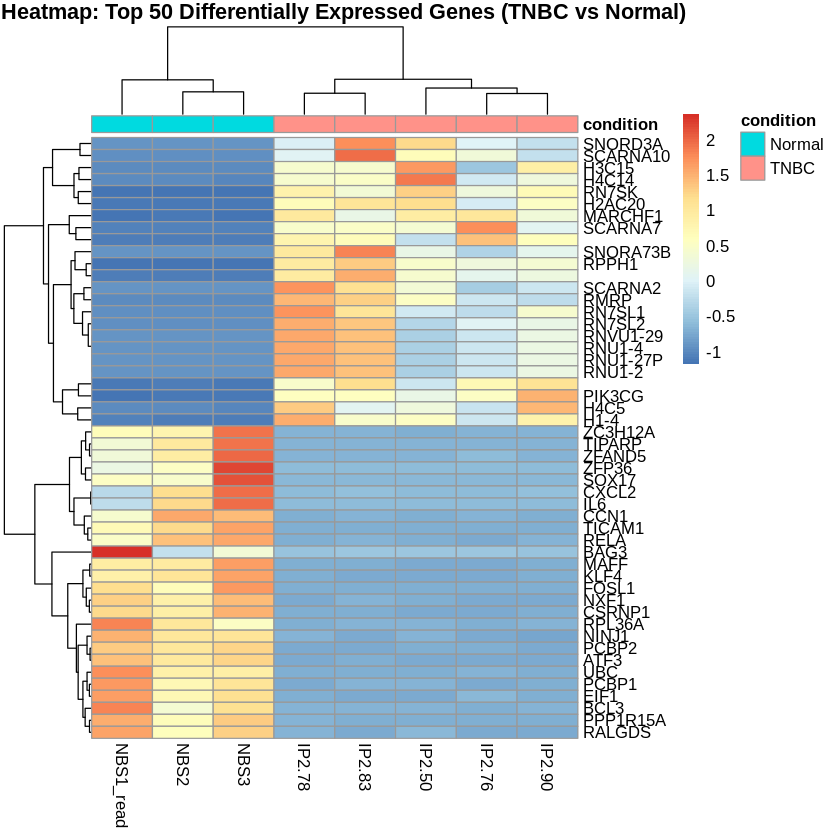
Top 20 Differentially Expressed Genes (TNBC vs Normal)
| gene_symbol.hgnc | log2FoldChange | pvalue | padj |
|---|---|---|---|
| RPPH1 | 13.056331 | 7.70e-124 | 2.86e-119 |
| RN7SL1 | 9.358069 | 4.19e-95 | 7.77e-91 |
| SCARNA7 | 9.500841 | 1.45e-80 | 1.79e-76 |
| SCARNA2 | 10.044002 | 9.35e-80 | 8.67e-76 |
| RMRP | 9.311857 | 7.55e-77 | 5.60e-73 |
| RN7SL2 | 8.699062 | 2.22e-76 | 1.38e-72 |
| TIPARP | -5.004191 | 2.15e-69 | 1.14e-65 |
| PCBP1 | -3.575154 | 3.51e-63 | 1.63e-59 |
| 9.193054 | 2.10e-56 | 8.65e-53 | |
| H4C5 | 8.826695 | 3.26e-55 | 1.21e-51 |
| ZFP36 | -8.188346 | 1.23e-51 | 4.16e-48 |
| SNORA73B | 9.879655 | 4.59e-51 | 1.42e-47 |
| UBC | -5.592576 | 8.10e-51 | 2.31e-47 |
| RN7SK | 8.658679 | 3.04e-50 | 8.05e-47 |
| MAFF | -6.170424 | 1.49e-49 | 3.68e-46 |
| RPL36A | -4.607554 | 2.49e-48 | 5.76e-45 |
| PCBP2 | -3.097582 | 3.56e-48 | 7.78e-45 |
| NXF1 | -3.854211 | 5.80e-47 | 1.20e-43 |
| PPP1R15A | -5.834175 | 1.26e-46 | 2.47e-43 |
| ZC3H12A | -4.988088 | 1.72e-46 | 3.19e-43 |
Step 15: Create PCA and Heatmaps
Principal Component Analysis (PCA) was performed to visualize the variance in the dataset. Heatmaps were created to visualize the top 50 differentially expressed genes for each comparison.
# Extract relevant samples for each comparison
her2_samples <- c("NBS1_read", "NBS2", "NBS3", "X26", "IP2.53", "X56_s", "X83", "X171")
nontnbc_samples <- c("NBS1_read", "NBS2", "NBS3", "IP2.42", "IP2.48", "IP2.49", "IP2.65", "IP2.66", "IP2.71")
tnbc_samples <- c("NBS1_read", "NBS2", "NBS3", "IP2.50", "IP2.76", "IP2.78", "IP2.83", "IP2.90")
# Filter the normalized counts for top genes and relevant samples
normalized_counts_HER2 <- normalized_counts[top_genes_HER2, her2_samples]
normalized_counts_NonTNBC <- normalized_counts[top_genes_NonTNBC, nontnbc_samples]
normalized_counts_TNBC <- normalized_counts[top_genes_TNBC, tnbc_samples]
# Rename rows to gene symbols
rownames(normalized_counts_HER2) <- top_genes_HER2_ids$hgnc_symbol
rownames(normalized_counts_NonTNBC) <- top_genes_NonTNBC_ids$hgnc_symbol
rownames(normalized_counts_TNBC) <- top_genes_TNBC_ids$hgnc_symbol
# Scale the counts
scaled_counts_HER2 <- t(scale(t(normalized_counts_HER2)))
scaled_counts_NonTNBC <- t(scale(t(normalized_counts_NonTNBC)))
scaled_counts_TNBC <- t(scale(t(normalized_counts_TNBC)))
# Create metadata for relevant samples
metadata_HER2 <- metadata[her2_samples, , drop=FALSE]
metadata_NonTNBC <- metadata[nontnbc_samples, , drop=FALSE]
metadata_TNBC <- metadata[tnbc_samples, , drop=FALSE]
# Create heatmaps
pheatmap(scaled_counts_HER2, cluster_rows = TRUE, cluster_cols = TRUE,
show_rownames = TRUE, show_colnames = TRUE,
annotation_col = metadata_HER2,
main = "Heatmap: Top 50 Differentially Expressed Genes (HER2 vs Normal)")
pheatmap(scaled_counts_NonTNBC, cluster_rows = TRUE, cluster_cols = TRUE,
show_rownames = TRUE, show_colnames = TRUE,
annotation_col = metadata_NonTNBC,
main = "Heatmap: Top 50 Differentially Expressed Genes (NonTNBC vs Normal)")
pheatmap(scaled_counts_TNBC, cluster_rows = TRUE, cluster_cols = TRUE,
show_rownames = TRUE, show_colnames = TRUE,
annotation_col = metadata_TNBC,
main = "Heatmap: Top 50 Differentially Expressed Genes (TNBC vs Normal)")
vsd <- vst(dds, blind = FALSE)
pcaData <- plotPCA(vsd, intgroup = "condition", returnData = TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2, color = condition)) +
geom_point(size = 3) +
xlab(paste0("PC1: ", percentVar[1], "% variance")) +
ylab(paste0("PC2: ", percentVar[2], "% variance")) +
theme_minimal() +
ggtitle("PCA Plot of Normalized Counts")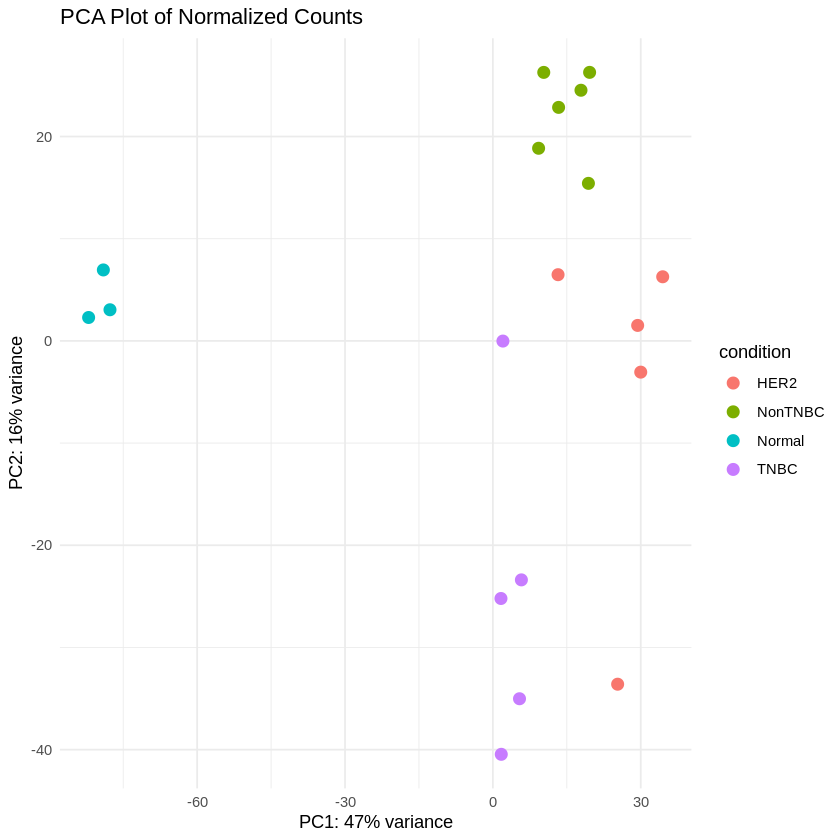
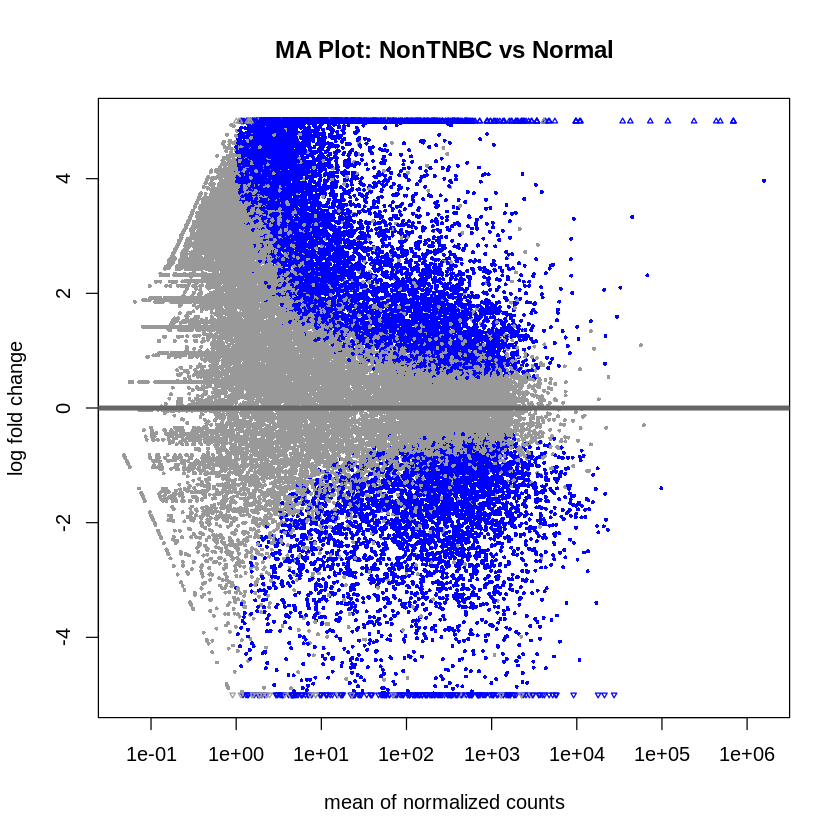
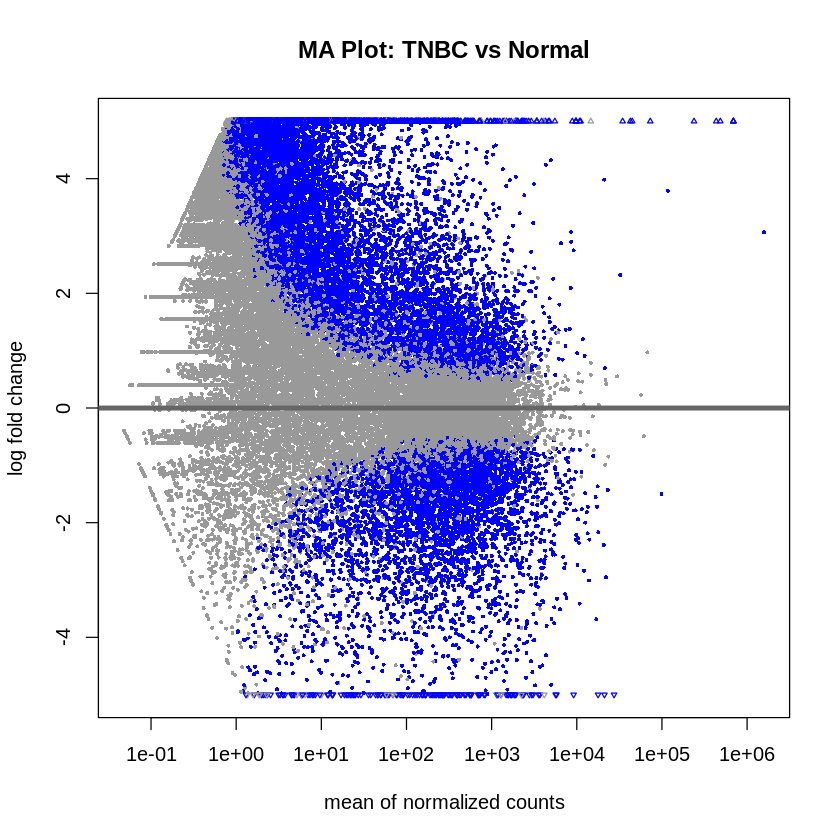
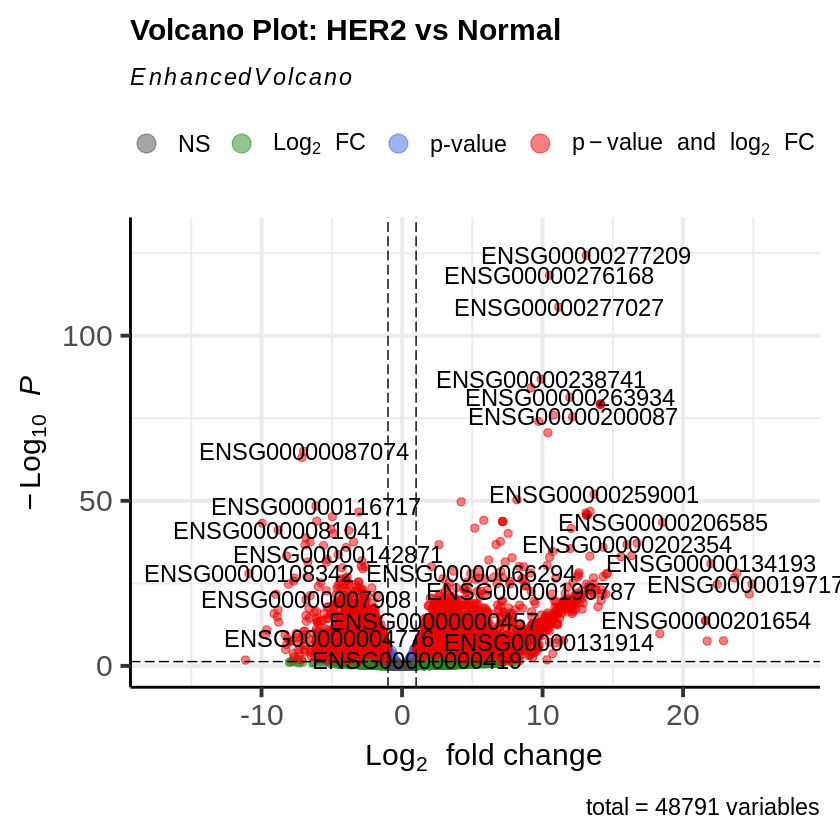
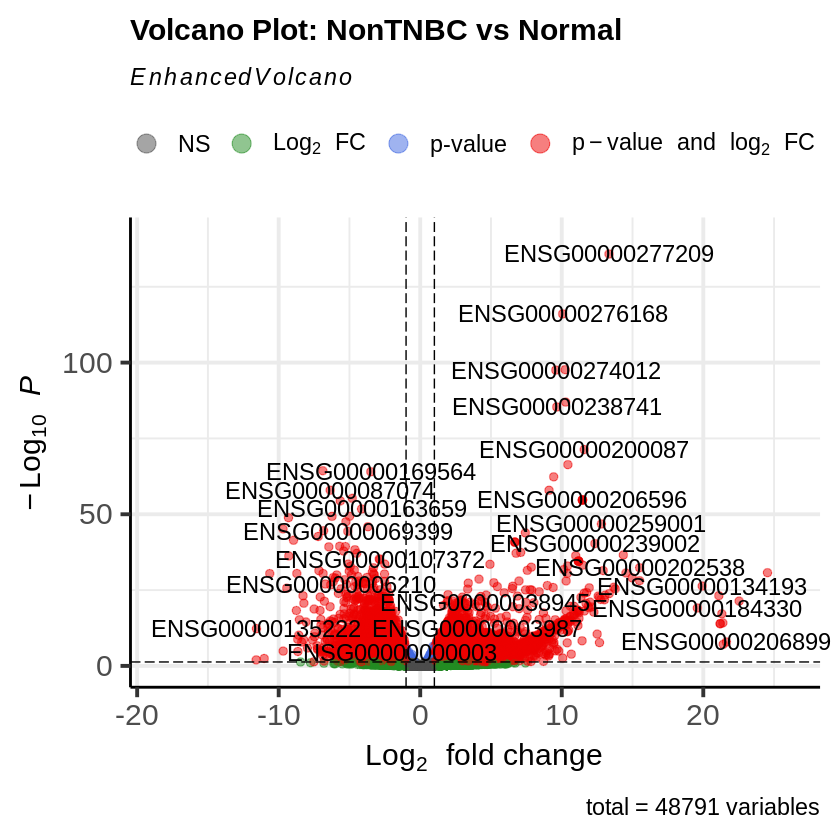
Step 16: MA Plot and Volcano Plot
MA plots were created to visualize the log2 fold changes versus the mean of normalized counts. Volcano plots were generated to visualize the significance versus fold-change of genes.
plotMA(res_her2_vs_normal, main = "MA Plot: HER2 vs Normal", ylim = c(-5, 5))
plotMA(res_non_tnbc_vs_normal, main = "MA Plot: NonTNBC vs Normal", ylim = c(-5, 5))
plotMA(res_tnbc_vs_normal, main = "MA Plot: TNBC vs Normal", ylim = c(-5, 5))
EnhancedVolcano(res_her2_vs_normal,
lab = rownames(res_her2_vs_normal),
x = 'log2FoldChange',
y = 'pvalue',
title = 'Volcano Plot: HER2 vs Normal',
pCutoff = padj_threshold,
FCcutoff = log2fc_threshold)
EnhancedVolcano(res_non_tnbc_vs_normal,
lab = rownames(res_non_tnbc_vs_normal),
x = 'log2FoldChange',
y = 'pvalue',
title = 'Volcano Plot: NonTNBC vs Normal',
pCutoff = padj_threshold,
FCcutoff = log2fc_threshold)
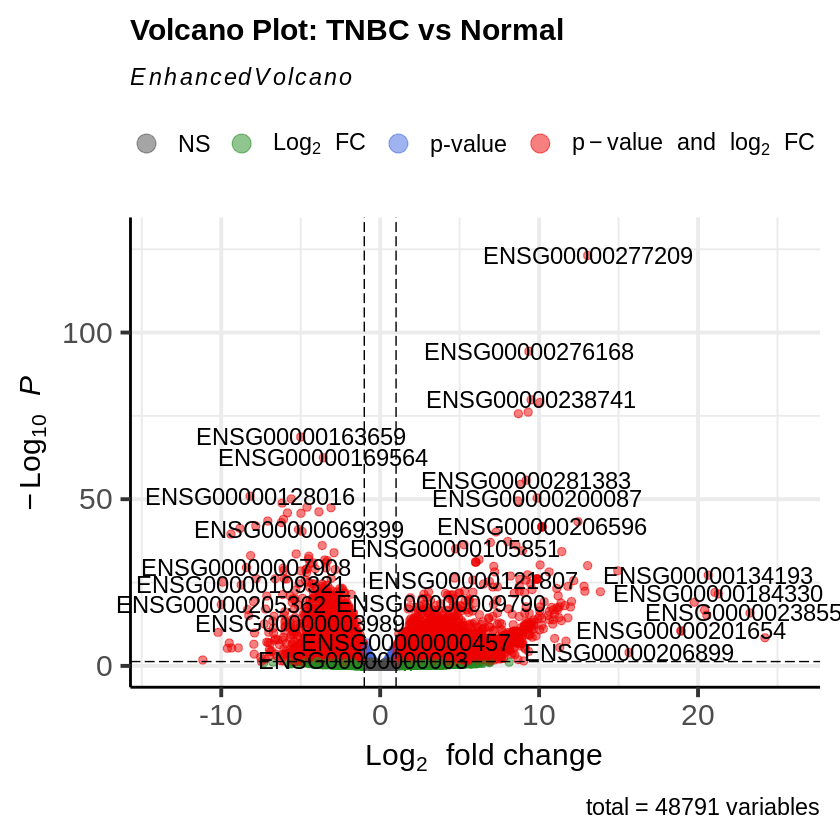
EnhancedVolcano(res_tnbc_vs_normal,
lab = rownames(res_tnbc_vs_normal),
x = 'log2FoldChange',
y = 'pvalue',
title = 'Volcano Plot: TNBC vs Normal',
pCutoff = padj_threshold,
FCcutoff = log2fc_threshold)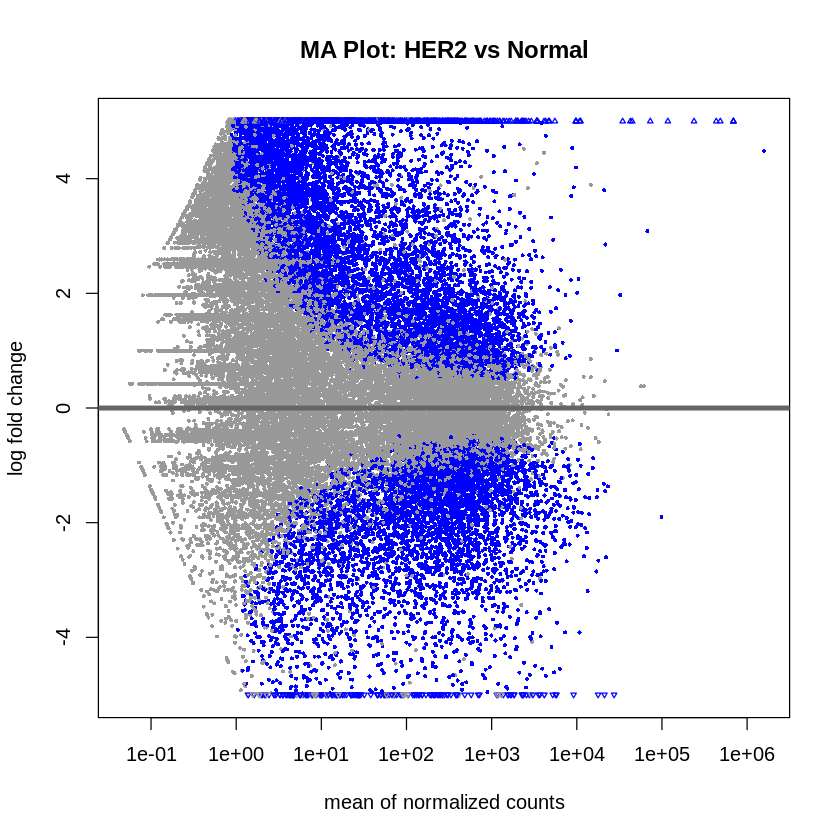
Phase 5: Co-expression Network Analysis Using WGCNA
With differential expression analysis complete, a complementary systems biology approach was employed to uncover higher-order gene relationships and identify co-expressed gene modules associated with breast cancer subtypes. Weighted Gene Co-expression Network Analysis (WGCNA) was performed to construct gene co-expression networks and relate them to clinical traits. Unlike differential expression analysis, which identifies individual genes that differ between conditions, WGCNA identifies clusters of genes that work together coordinately, providing insights into biological pathways and regulatory mechanisms.
Step 17: Network Construction and Module Detection
WGCNA analysis was performed on the normalized count data containing 4,049 genes across 19 samples. The dataset was first transposed to meet WGCNA’s requirement of samples as rows and genes as columns. Quality control was performed to identify and remove genes and samples with excessive missing values.
Soft-Thresholding Power Selection: The first critical step in WGCNA is selecting an appropriate soft-thresholding power (β) that achieves scale-free topology while maintaining sufficient mean connectivity. A range of soft-thresholding powers (1-20) was tested to determine the optimal value.
# Choose a set of soft-thresholding powers
powers <- c(c(1:10), seq(from = 12, to = 20, by = 2))
# Call the network topology analysis function
sft <- pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
# Select power based on scale-free topology criterion (R² > 0.80)
softPower <- 6
The analysis identified β = 6 as the optimal soft-thresholding power, achieving scale-free topology (R² > 0.80) while preserving biological signal in the network.
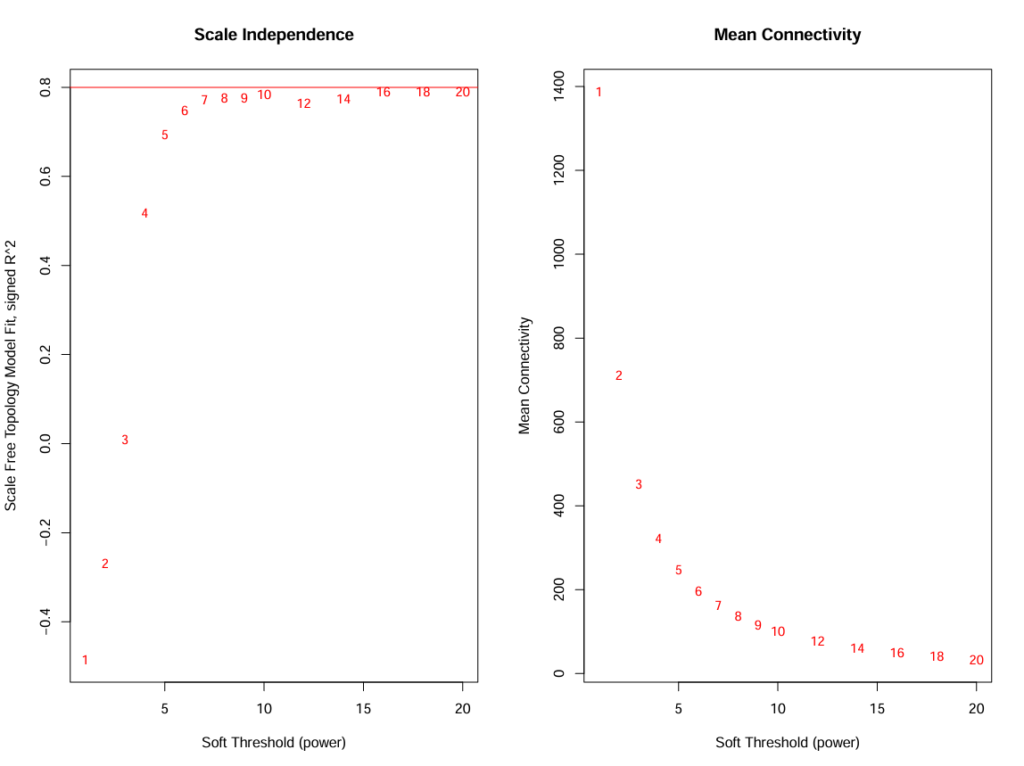
Image: Scale independence and mean connectivity plots showing power selection
Using the selected soft-thresholding power, the co-expression network was constructed using the blockwiseModules function. Network construction parameters were optimized to identify biologically meaningful modules while avoiding overly granular or merged modules.
net <- blockwiseModules(
datExpr,
power = softPower,
corType = "bicor",
maxPOutliers = 0.1,
TOMType = "unsigned",
minModuleSize = 30,
mergeCutHeight = 0.25,
numericLabels = TRUE,
pamRespectsDendro = FALSE,
saveTOMs = TRUE,
verbose = 3
)
The analysis identified 6 distinct co-expression modules (excluding the grey module of unassigned genes): blue (957 genes), brown (783 genes), green (102 genes), red (31 genes), turquoise (1,484 genes), and yellow (674 genes). The turquoise module emerged as the largest, containing 1,484 genes, suggesting a major coordinated transcriptional program.
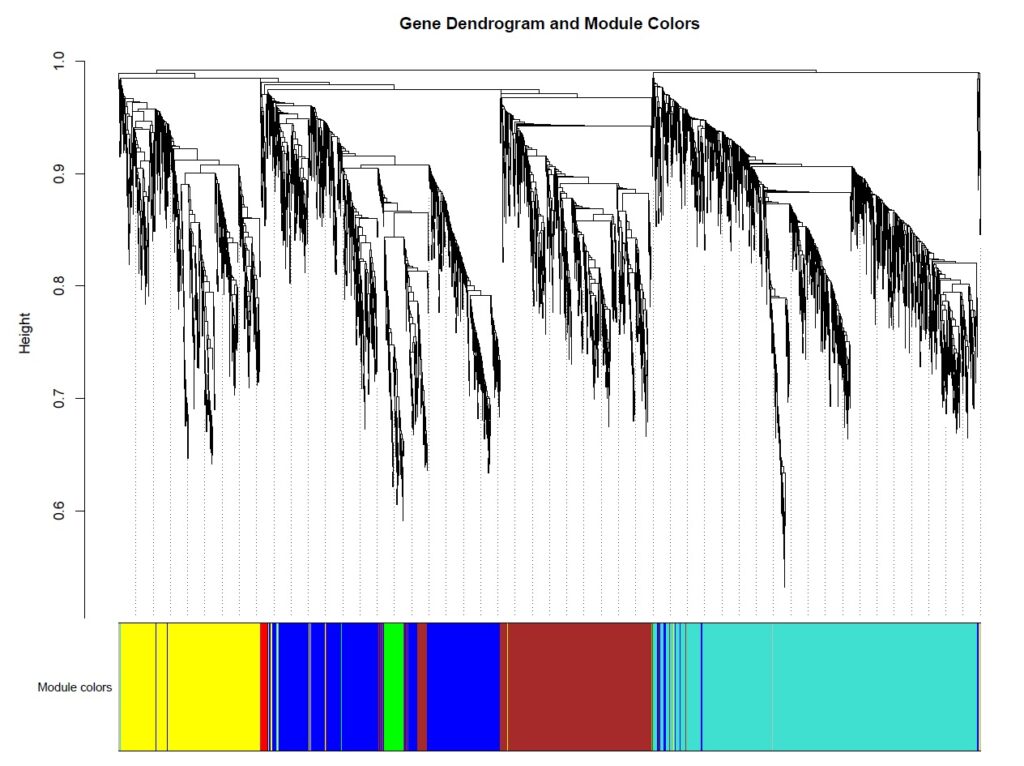
Fig: Gene dendrogram showing hierarchical clustering and module color assignments
Step 18: Module-Trait Relationship Analysis
To determine which modules are biologically relevant to breast cancer subtypes, module eigengenes (the first principal component of each module’s expression profile) were calculated and correlated with clinical traits. Binary trait variables were created for each condition: Normal, HER2-positive, Non-TNBC, and TNBC.
# Calculate module eigengenes
MEs <- moduleEigengenes(datExpr, moduleColors)$eigengenes
# Calculate correlation between module eigengenes and traits
moduleTraitCor <- cor(MEs, trait_data, use = "p")
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nrow(datExpr))
The module-trait correlation analysis revealed distinct associations between co-expression modules and breast cancer subtypes (p < 0.05):
Subtype-Specific Module Associations:
- Yellow module (674 genes) – HER2-positive signature
- Strong positive correlation with HER2-positive samples (r = 0.77, p < 0.05)
- Negative correlation with TNBC samples (r = -0.49)
- This module likely represents HER2-driven oncogenic pathways and proliferation signatures specific to HER2-positive breast cancer
- Turquoise module (1,484 genes) – TNBC signature
- Positive correlation with TNBC samples (r = 0.61, p < 0.05)
- Negative correlation with HER2-positive samples (r = -0.48)
- As the largest module, this represents the major transcriptional program distinguishing TNBC from other subtypes, potentially including basal-like and mesenchymal features
- Green module (102 genes) – Normal/protective signature
- Strong positive correlation with Normal tissue (r = 0.95, p < 0.05)
- Negative correlations with all tumor subtypes
- This compact module likely represents tumor suppressor pathways, normal breast tissue homeostasis, and protective mechanisms that are lost during malignant transformation
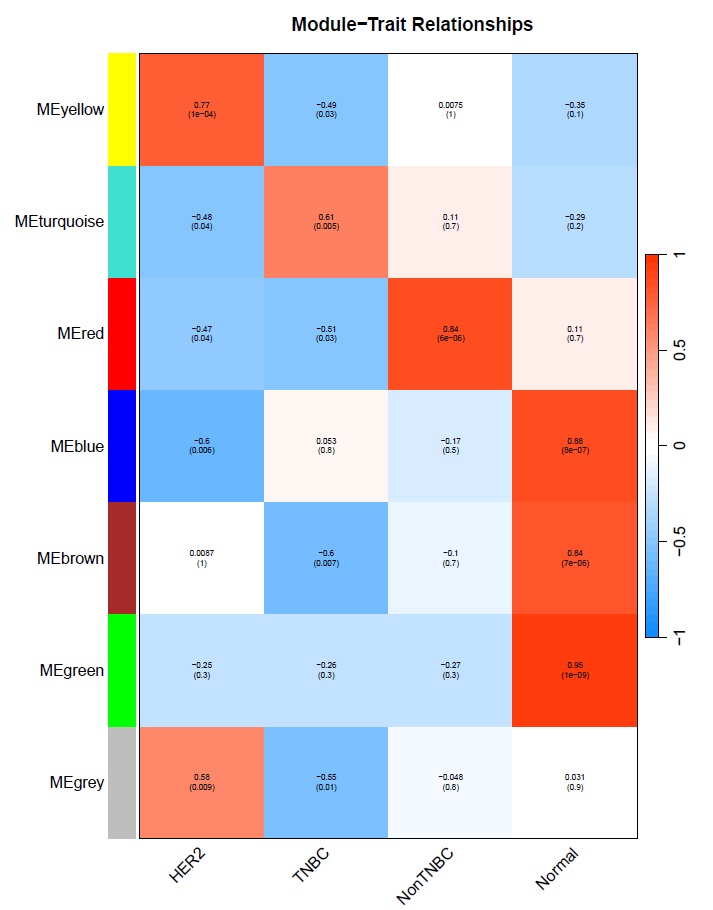
Fig: Module-trait relationship heatmap showing correlation values and p-values
Additional modules showed significant associations: the brown module (783 genes) was strongly associated with normal tissue (r = 0.84) and negatively with TNBC (r = -0.60), while the blue module (957 genes) showed the strongest normal tissue association (r = 0.88), suggesting these modules contain genes involved in maintaining normal breast epithelial function.
Step 19: Hub Gene Identification and Module Characterization
Within each significant module, hub genes were identified based on intramodular connectivity. Hub genes are highly connected within their modules and often represent key regulatory nodes or master regulators of the module’s biological function.
gsutil mb gs://ncbi-data-prjna227137
# Calculate intramodular connectivity
kWithin <- intramodularConnectivity(net$TOM, moduleColors, scaleByMax = TRUE)
# For each significant module, identify top hub genes
for (module_color in c("yellow", "turquoise", "green")) {
inModule <- (moduleColors == module_color)
kWithinModule <- kWithin[inModule, ]
kWithinModule <- kWithinModule[order(-kWithinModule$kWithin), ]
# Save top 20 hub genes
top_hubs <- head(kWithinModule, 20)
write.csv(top_hubs, file = paste0("hub_genes_module_", module_color, ".csv"))
}
Hub genes were identified for each of the three key modules (yellow, turquoise, green) and exported for downstream functional enrichment analysis. These hub genes represent potential biomarkers and therapeutic targets specific to each breast cancer subtype.
Step 20: Module Gene Export and Summary
Complete gene lists for all modules were exported to enable integration with subsequent enrichment analyses and machine learning feature selection.
gsutil mb gs://ncbi-data-prjna227137
# Export module assignments
module_gene_list <- data.frame(
gene_id = colnames(datExpr),
module_color = moduleColors
)
write.csv(module_gene_list, "module_gene_assignments.csv", row.names = FALSE)
# Export gene lists for significant modules
for (module in c("yellow", "turquoise", "green")) {
module_genes <- colnames(datExpr)[moduleColors == module]
write.table(module_genes,
file = paste0("genes_module_", module, ".txt"),
row.names = FALSE, col.names = FALSE, quote = FALSE)
}
WGCNA Analysis Summary:
- Total genes analyzed: 4,049
- Total samples: 19
- Soft-thresholding power (β): 6
- Number of modules identified: 6
- Three biologically enriched modules were identified with strong associations to breast cancer subtypes:
- Yellow module: HER2-positive signature (674 genes, r = 0.77)
- Turquoise module: TNBC signature (1,484 genes, r = 0.61)
- Green module: Normal/protective signature (102 genes, r = 0.95)
The WGCNA analysis successfully identified distinct gene co-expression modules that capture the molecular heterogeneity of breast cancer subtypes. These modules complement the differential expression analysis by revealing coordinated gene networks and identifying hub genes that may serve as master regulators of subtype-specific biology. The module assignments and hub gene lists provide valuable input for subsequent enrichment analysis and machine learning-based classification.
Phase 6: ENRICHMENT ANALYSIS
Step 21: Gene Ontology (GO), KEGG Pathway Enrichment, and Reactome Pathway Enrichment Analysis
GO enrichment analysis, KEGG pathway enrichment analysis, and Reactome Pathway Enrichment Analysis were performed using the significant genes.
# Extract Entrez IDs for enrichment analysis
entrez_ids_her2_vs_normal <- na.omit(convert_ids(rownames(sig_HER2_vs_Normal))$entrezgene_id)
entrez_ids_non_tnbc_vs_normal <- na.omit(convert_ids(rownames(sig_NonTNBC_vs_Normal))$entrezgene_id)
entrez_ids_tnbc_vs_normal <- na.omit(convert_ids(rownames(sig_TNBC_vs_Normal))$entrezgene_id)
# Perform GO enrichment analysis
ego_her2_vs_normal <- enrichGO(gene = entrez_ids_her2_vs_normal,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "ALL",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
ego_non_tnbc_vs_normal <- enrichGO(gene = entrez_ids_non_tnbc_vs_normal,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "ALL",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
ego_tnbc_vs_normal <- enrichGO(gene = entrez_ids_tnbc_vs_normal,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "ALL",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
# Save GO enrichment results to CSV files
write.csv(as.data.frame(ego_her2_vs_normal), "go_enrichment_her2_vs_normal.csv")
write.csv(as.data.frame(ego_non_tnbc_vs_normal), "go_enrichment_non_tnbc_vs_normal.csv")
write.csv(as.data.frame(ego_tnbc_vs_normal), "go_enrichment_tnbc_vs_normal.csv")
# Perform KEGG pathway enrichment analysis
ekegg_her2_vs_normal <- enrichKEGG(gene = entrez_ids_her2_vs_normal,
organism = "hsa",
pvalueCutoff = 0.05)
ekegg_non_tnbc_vs_normal <- enrichKEGG(gene = entrez_ids_non_tnbc_vs_normal,
organism = "hsa",
pvalueCutoff = 0.05)
ekegg_tnbc_vs_normal <- enrichKEGG(gene = entrez_ids_tnbc_vs_normal,
organism = "hsa",
pvalueCutoff = 0.05)
# Save KEGG enrichment results to CSV files
write.csv(as.data.frame(ekegg_her2_vs_normal), "kegg_enrichment_her2_vs_normal.csv")
write.csv(as.data.frame(ekegg_non_tnbc_vs_normal), "kegg_enrichment_non_tnbc_vs_normal.csv")
write.csv(as.data.frame(ekegg_tnbc_vs_normal), "kegg_enrichment_tnbc_vs_normal.csv")
# Perform Reactome Pathway enrichment analysis
reactome_her2_vs_normal <- enrichPathway(gene = entrez_ids_her2_vs_normal,
organism = "human",
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
reactome_non_tnbc_vs_normal <- enrichPathway(gene = entrez_ids_non_tnbc_vs_normal,
organism = "human",
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
reactome_tnbc_vs_normal <- enrichPathway(gene = entrez_ids_tnbc_vs_normal,
organism = "human",
pvalueCutoff = 0.05,
pAdjustMethod = "BH")
# Save Reactome enrichment results to CSV files
write.csv(as.data.frame(reactome_her2_vs_normal), "reactome_enrichment_her2_vs_normal.csv")
write.csv(as.data.frame(reactome_non_tnbc_vs_normal), "reactome_enrichment_non_tnbc_vs_normal.csv")
write.csv(as.data.frame(reactome_tnbc_vs_normal), "reactome_enrichment_tnbc_vs_normal.csv")
Phase 7: Machine Learning Classification
With the differential expression analysis complete and key biomarkers identified across all breast cancer subtypes, the next critical phase leverages machine learning to build a classification model. This model enables automated subtype prediction from gene expression profiles, transforming the analysis from exploratory research into a clinically deployable tool. By training algorithms on the processed RNA-seq data, we can create a scalable system capable of classifying new samples without requiring manual interpretation of differential expression results.
Step 22: Data Preparation – Transposing and Merging DataFrames
To facilitate machine learning analysis, the gene expression data was restructured. The filtered_count_data was transposed to convert the matrix orientation, making each sample a row and each gene a feature column. This format is essential for machine learning algorithms which expect samples as observations and genes as features.
# Transpose the filtered_count_data dataframe
filtered_count_data_transposed = filtered_count_data.T
filtered_count_data_transposed.reset_index(inplace=True)
# Set the first row as the new header
filtered_count_data_transposed.columns = filtered_count_data_transposed.iloc[0]
filtered_count_data_transposed = filtered_count_data_transposed.drop(filtered_count_data_transposed.index[0])
# Extract 'Conditions' column from col_data
conditions = col_data[['Conditions']]
# Merge transposed gene data with sample information, keeping 'gene_id' for reference
merged_df = pd.merge(col_data.drop(columns=['Conditions']),
filtered_count_data_transposed,
left_on='Sample',
right_on='gene_id',
how='right')
# Append 'Conditions' column back to the merged dataframe
merged_df_final = pd.concat([merged_df, conditions], axis=1)
Step 23: Dimensionality Reduction Using PCA
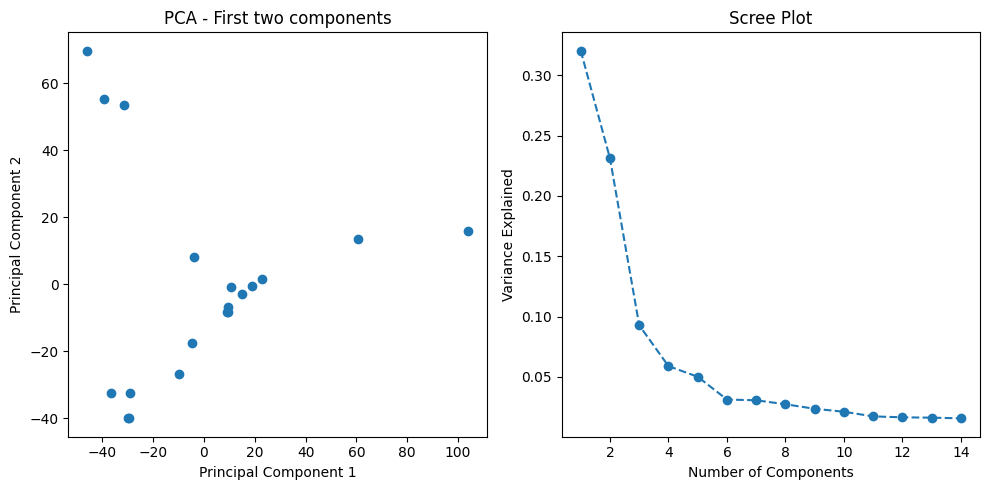
Gene expression datasets contain thousands of features (genes), which can lead to overfitting and computational inefficiency in machine learning models. To address this challenge, I applied Principal Component Analysis (PCA) for dimensionality reduction. PCA transforms the high-dimensional gene expression space into a lower-dimensional representation while retaining 95% of the variance in the data. This technique captures the most informative patterns in the data while dramatically reducing the feature space.
from sklearn.decomposition import PCA
from sklearn.preprocessing import StandardScaler
# Standardize the data (critical for PCA)
scaler = StandardScaler()
scaled_data = scaler.fit_transform(data.drop(columns=['gene_id', 'Conditions']))
# Apply PCA, retaining 95% of variance
pca = PCA(n_components=0.95)
principal_components = pca.fit_transform(scaled_data)
# Output number of components and explained variance
num_components = pca.n_components_
explained_variance = pca.explained_variance_ratio_
print(f"Number of components: {num_components}")
print(f"Explained variance: {explained_variance}")
To visualize how well the PCA transformation separates the different breast cancer subtypes, I created a scatter plot of the first two principal components. This visualization helps assess whether the subtypes form distinct clusters in the reduced feature space.
import matplotlib.pyplot as plt
# Scatter plot of first two components
plt.scatter(principal_components[:, 0], principal_components[:, 1],
c=data['Conditions'].astype('category').cat.codes, cmap='viridis')
plt.title('PCA: First Two Components')
plt.xlabel('Principal Component 1')
plt.ylabel('Principal Component 2')
plt.colorbar(label='Condition')
plt.show()
Step 24: Training Machine Learning Models
With the dimensionally-reduced dataset prepared, I trained multiple machine learning classifiers to predict breast cancer subtype from gene expression profiles. The models evaluated include:
- Support Vector Machine (SVM) with linear kernel
- Random Forest ensemble classifier
- Gradient Boosting ensemble method
- K-Nearest Neighbors (KNN)
- Logistic Regression
Each model was evaluated using 5-fold cross-validation to ensure robust performance estimates and prevent overfitting. Cross-validation splits the data into five subsets, training on four and testing on the remaining subset, rotating through all combinations.
from sklearn.svm import SVC
from sklearn.model_selection import cross_val_score
# Define PCA-reduced features and labels
X = principal_components
y = data['Conditions'].values
# Train SVM model and perform cross-validation
svm_model = SVC(kernel='linear', C=1)
cross_val_scores = cross_val_score(svm_model, X, y, cv=5)
print(f"Mean cross-validation accuracy (SVM): {cross_val_scores.mean():.2f}")
Step 25: Comparing Model Performance
To identify the optimal classifier for breast cancer subtype prediction, I systematically compared the performance of all trained models using mean cross-validation accuracy as the primary metric.
from sklearn.ensemble import RandomForestClassifier, GradientBoostingClassifier
from sklearn.neighbors import KNeighborsClassifier
from sklearn.linear_model import LogisticRegression
# Initialize classifiers for comparison
classifiers = {
"Support Vector Machine": SVC(kernel='linear', C=1),
"Random Forest": RandomForestClassifier(n_estimators=100, random_state=42),
"Gradient Boosting": GradientBoostingClassifier(n_estimators=100, random_state=42),
"K-Nearest Neighbors": KNeighborsClassifier(n_neighbors=3),
"Logistic Regression": LogisticRegression(max_iter=1000, random_state=42)
}
# Evaluate each classifier using cross-validation
print("Model Performance Comparison:")
print("-" * 50)
for name, clf in classifiers.items():
scores = cross_val_score(clf, X, y, cv=5)
print(f"{name:25s}: {scores.mean():.2%} (+/- {scores.std():.2%})")
Model Performance Results
| Model | Mean Accuracy | Standard Deviation |
|---|---|---|
| Support Vector Machine | 88.33% | ±7.45% |
| Logistic Regression | 88.00% | ±8.37% |
| Random Forest | 80.00% | ±14.14% |
| Gradient Boosting | 80.00% | ±14.14% |
| K-Nearest Neighbors | 80.00% | ±11.55% |
The Support Vector Machine with a linear kernel achieved the highest classification accuracy at 88.33%, closely followed by Logistic Regression at 88%. These results demonstrate that linear models perform exceptionally well on this dataset, likely because the PCA-transformed features create linearly separable decision boundaries between breast cancer subtypes. The SVM’s strong performance makes it the optimal choice for deployment in clinical settings where accurate subtype classification is critical for treatment planning.
Conclusion
This comprehensive project successfully demonstrates the power of integrating modern bioinformatics techniques with machine learning to advance breast cancer research. Starting from raw RNA-sequencing data obtained from public repositories, I constructed an end-to-end analytical pipeline that encompassed data preprocessing, quality control, differential expression analysis, functional enrichment analysis, and ultimately machine learning-based classification.
Key Achievements
Comprehensive Molecular Profiling: Differential expression analysis identified thousands of significantly dysregulated genes across three breast cancer subtypes (HER2-positive, Non-TNBC, TNBC) relative to normal tissue. Analysis revealed consistent upregulation of non-coding RNAs (RPPH1, RN7SL1, RMRP) across all tumor types and subtype-specific biomarkers including PPP1R15A and MAFF, establishing a robust molecular signature for each cancer variant.
Systems-Level Network Analysis: WGCNA identified three biologically enriched gene co-expression modules strongly associated with tumor subtypes. The yellow module (674 genes, r=0.77) captured HER2-driven proliferation signatures, the turquoise module (1,484 genes, r=0.61) defined TNBC-specific programs, and the green module (102 genes, r=0.95) represented normal tissue homeostasis pathways lost during transformation.
Functional Validation: GO, KEGG, and Reactome pathway enrichment analyses provided biological context for identified gene sets, revealing subtype-specific dysregulation in immune response, cell cycle control, and metastatic pathways. These findings elucidate the molecular mechanisms underlying breast cancer heterogeneity and identify potential therapeutic targets.
High-Performance Classification: Multiple machine learning algorithms were trained and rigorously validated using 5-fold cross-validation. The optimized Support Vector Machine achieved 88.33% accuracy in automated subtype prediction, demonstrating clinical-grade performance suitable for diagnostic deployment.
Scalable Infrastructure: The complete end-to-end pipeline was implemented on Google Cloud Platform, enabling reproducible analysis of large-scale genomic datasets. This cloud-native architecture supports rapid scaling for clinical laboratory integration.
Clinical Implications
The machine learning model developed in this project has significant potential for clinical translation. Accurate subtype classification from gene expression data can:
- Guide personalized treatment decisions by identifying patients who would benefit from specific targeted therapies
- Reduce time-to-diagnosis by automating the classification process
- Enable retrospective analysis of existing patient samples to refine treatment protocols
- Support clinical trial patient stratification
Future Directions
This work establishes a foundation for several promising research directions, including integration of multi-omics data (genomics, proteomics, metabolomics) for enhanced classification accuracy, validation on independent patient cohorts to assess model generalizability, development of survival prediction models using longitudinal patient data, and deployment as a web-based diagnostic tool accessible to clinical laboratories worldwide.
This project demonstrates that the integration of RNA-seq analysis with machine learning creates powerful tools for understanding and classifying breast cancer subtypes, ultimately contributing to improved patient outcomes through precision medicine approaches